FastQC
用途
快速對產出的NGS資料做QC...
使用方法
FastQC是java所寫成的,所以安裝之前要確認系統有安裝JVM。
FastQC可以吃的檔案格式有:
- .fastq
- .fastq.gz
- .SAM/.BAM
- 其他...(說明)
而一般NGS的data出來就已經打包成fast.gz檔了,直接用gz餵給fastQC就好,指令長得像這樣:
fastqc filename.fastq.gz
分析完成之後會產生一個html檔、一個zip檔,可以從server端下載html檔打開看整份報告:
filename_fastqc.html
filename_fastqc.zip
報告解讀
詳細的官方版本可以參考這裡。
出來的報告有許多項目,會以icon標記三種狀態,告訴你每個項目的QC有沒有通過:
 PASS
PASS  WARNING
WARNING  FAIL
FAIL
Basic Statistics
列一些基本資訊,這個地方不會報錯
| Measure | Value |
|---|---|
| Filename #顯示丟給fastqc處裡的檔案名稱 | example_file.fastq.gz |
| File type #Actual base calls or colorspace data which had to be converted to base calls (不會翻先暫時這樣) | Conventional base calls |
| Encoding #列出機器版本,讓人判斷編碼方式 | Sanger / Illumina 1.9 |
| Total Sequences #總reads數 | 18000000 |
| Sequences flagged as poor quality | 0 |
| Sequence length #每個read的讀長 | 110 |
| %GC #The overall GC Content(%) | 50 |
Per base sequence quality
說明:FastQ檔案對於每個位置的序列品質劃出box plot。
- X軸:base的位置。
- Y軸:品質分數。
- 箱型圖:
- 中心紅線:中位數(Median)。
- 黃色箱型:Q1(25%)-Q3(75%)。
- 上下底線:10%、90% 所在分數。
- 藍色線:平均值(Mean)。
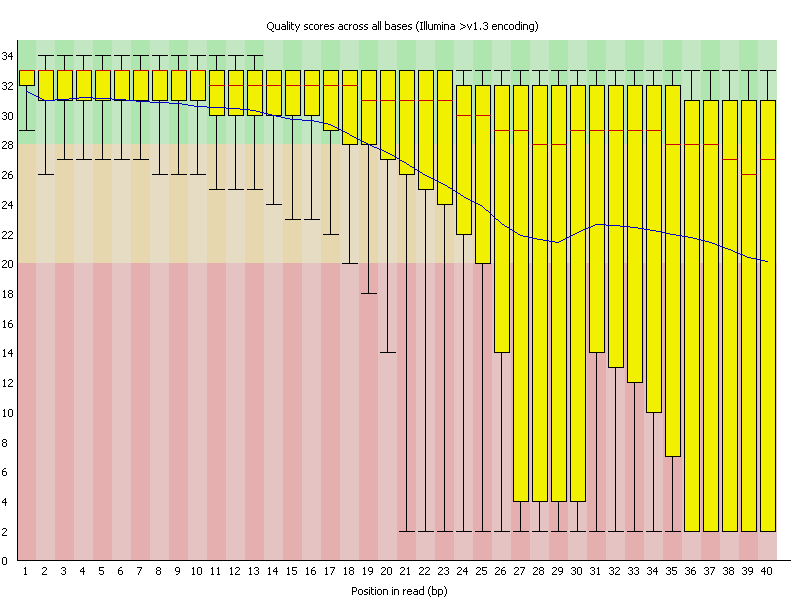
可以看到愈遠的位置讀到的錯誤率愈高,所以定序在末端的位置分數分度愈高愈好,在品質不好的位置在後續分析時應考慮去除。
Per tile sequence quality
好像有點深奧用處又不是很常會看,先欠著...
Per sequence quality scores
說明:用來檢查是否有某些read分數特別低。
- X軸:分數。
- Y軸:得到某分數的read數。
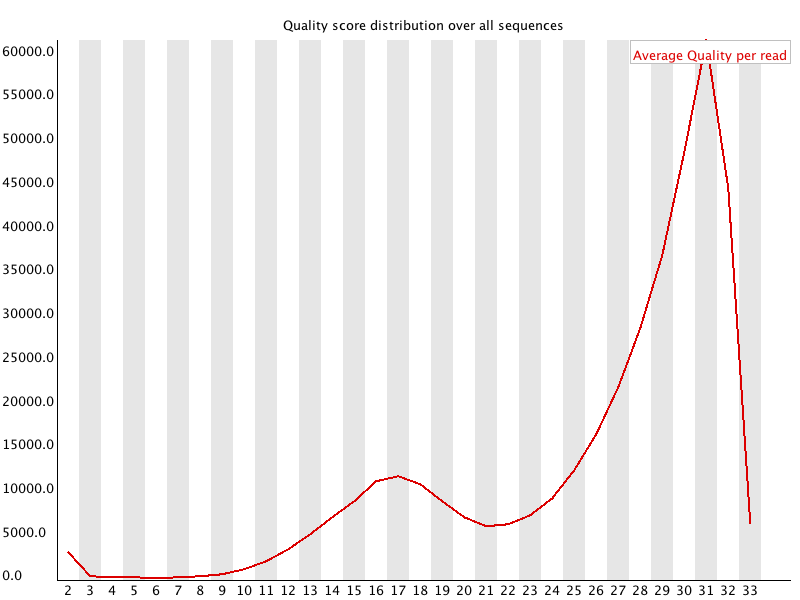
正常來說會有一群分數比較低的read,是因為flow cell的邊緣照相品質特別不好因此會得到較差的分數,但是邊緣的read佔總體的百分比理論上要很少。但如果定序的結果總體來講分數不高,那就需要考慮是否由一些其他系統性的問題所造成。
異常:
- Warning:the most frequently observed mean quality < 27 (~=0.2% error rate).
- Failure:the most frequently observed mean quality < 20 (~=1% error rate).
P.S.如果餵給fastqc的檔案是.SAM.BAM,由於檔案當中沒有紀錄品質分數相關資訊,所以這個module的分析結果不會被顯示出來。
PS:經過Trimmomatic之類的軟體修剪後的檔案,無法看出原本定序的Per sequence quality scores。
P.S.Common reasons for warnings這一段看不懂。
Per base sequence content
說明:用來檢查每個位置ATCG是否分布平均。
- X軸:位置。
- Y軸:ATCG的Content(%)。
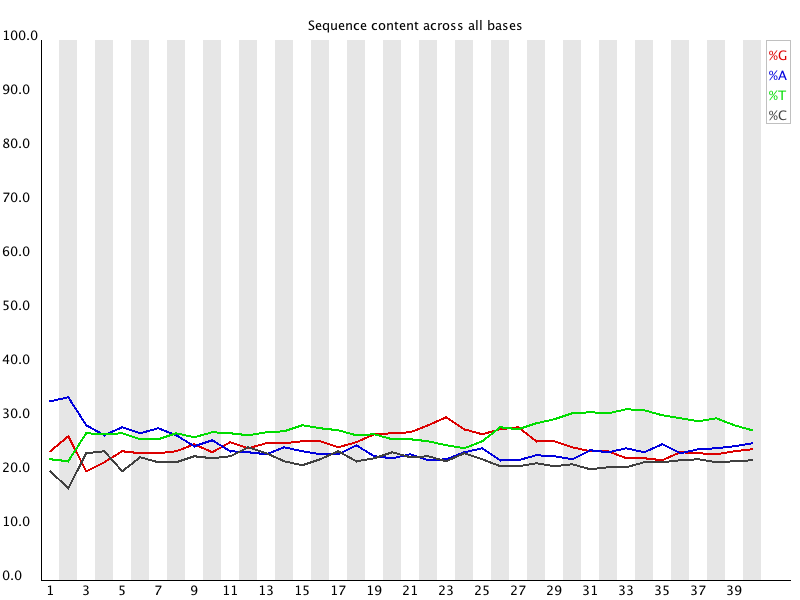
以一個隨機的library做出來的資料來說,因為斷點會隨機發生,所以每一個位置的ATCG比例應該和總體定序的ATCG比例是一樣的,也就是說ATCG四條線最完美的情況是相互平行。
某些library在開始的位置會有雜亂現象,像是:
- priming使用到random hexamers(幾乎所有RNA-seq的製備都會使用)
- fragmentation過程中使用transposases
這些技術性造成的偏差無法藉由trimming之類的技術消除掉,但對下游分析的影響也不大。//還有一段Kmer什麼的看不懂。
異常:
- Warning:任何位置ATCG之間的比例差異超過10%。
- Failure:任何位置ATCG之間的比例差異超過20%。
可能造成異常的原因:- Overrepresented sequence
adapter dimers 或 rRNA。 - Biases Fragmentation
接上random hexamers或用tagmentation產生的library理論上序列會分布平均,但是經驗上告訴我們在前12bp左右會有selection bias的情況,幾乎所有做RNA-Seq的library都會因為上述原因造成這個module報錯,但是實際上經過後續的trimming就不會影像後續的mapping跟expression measuring。 - Biases Composition Libraries
有些library天生就長得跟別人不一樣,例如要分析methylation的樣本會加入sodium bisulfite將未甲基化的C轉成T,這時候序列的ATCG含量就不會一樣(C會特別少),儘管在這種library是很正常的現象仍然觸發這個module報錯。 - Extreme Trimming
有時候因為要將堆疊在一起的adaptor去掉,而將adaptor相似的非adaptor序列也殺乾淨了,會在尾端產生假的bias(業障重啊...)。
- Overrepresented sequence
Per sequence GC content
說明:用來檢查樣本每個read的CG%,是否符合常態分布曲線的理論值。
- X軸:每個read的GC%
- Y軸:擁有特定GC%的read數目。
- 藍色線:母群體參考分布曲線。
- 紅色線:定序的結果。
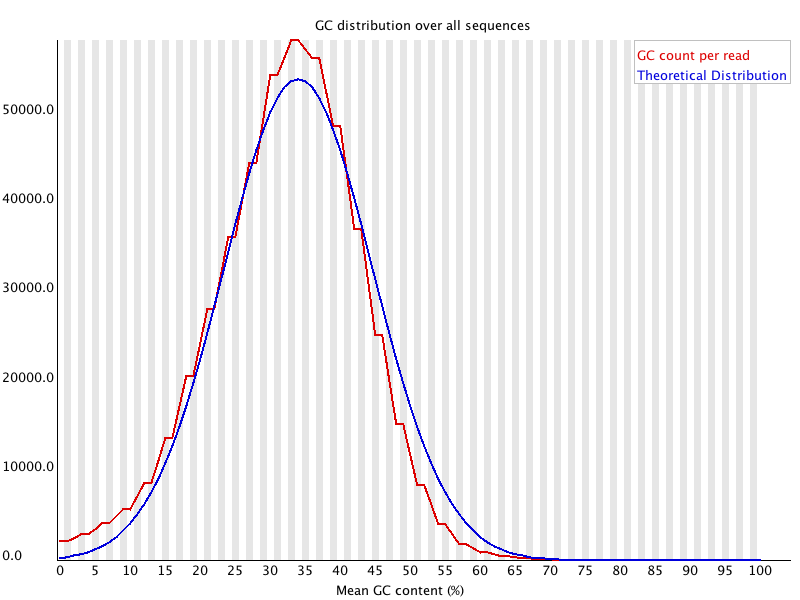
拿WGS來說,如果library做得夠平均讀出來的GC content分布應該跟整個Ganome的分布情況相同(從母群體中作大樣本隨機取樣的概念),但是由於fastqc不知道整個genome的GC含量分布情況,所以是從輸入檔案去取得相關統計數據(mean、standard error...)回推母群體,再畫出藍色的母群體參考分布曲線。
異常:
- 曲線線型異常
- Adaptor Contamination
在樣本中增加許多帶有特定GC%的某族群,會使波形變窄或雙峰分布。 - Coss-species Contamination
將GC%分布情形不同的各樣本混在一起,會導致波形變寬或雙峰分布。 - 其他種類的原因。
- Adaptor Contamination
- 曲線為常態分布但中心偏離理論值
可能為系統性誤差(取樣偏差)所致,這個情況就不會跳異常,因為fastqc是拿樣本的GC%去推測分布曲線應該長什麼樣子(拿偏差的取樣假設為隨機取樣,推測樣本母群體的分布情形),再去檢查實際情形是否符合,不會知道樣本的GC%理論上(母群體實際上的分布曲線)應該是多少。 - Warning
常態分布區線的sum of the deviations (sum(Xi-Xmean))與樣本的sum of the deviations,相差超過15%。 - Failure
同上,超過30%。
Per base N content
說明:顯示在所有read的某個位置,N含量的比例。
//P.S.如果定序時無法判斷某個點是何種鹼基,會用N代替。
- X軸:序列位置
- Y軸:所有read在某個位置N的含量(%)。
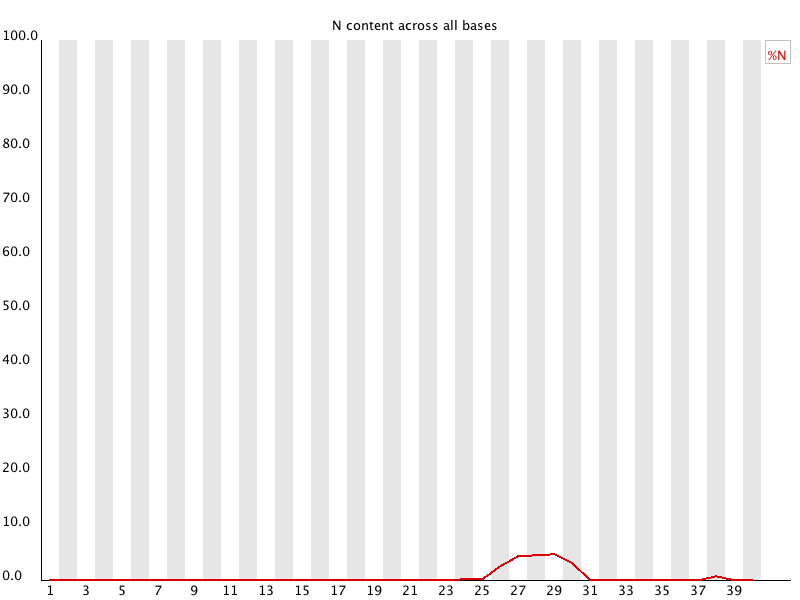 N的含量(%)應該都要很低(尤其是在考進序列的尾端)。
如果N含量稍微高一些,代表後須分析無法從data解讀出有效的base call.
N的含量(%)應該都要很低(尤其是在考進序列的尾端)。
如果N含量稍微高一些,代表後須分析無法從data解讀出有效的base call.
異常:
- Warning:有一個以上的位置N含量>5%。
- Failure:有一個以上的位置N含量>20%。
- Common reasons
- general loss of quality
如果N%偏高一般代表定序品質不良,所以最好跟其他的qc module作系統性審視,可以挑各個N%較高的區塊(ex:25-31)去檢查coverage的情形,在這個case裡面尾端N%較高的區塊可能含有很少的序列(coverage偏低),會觸發某些error。 - high proportions of N at a small number of positions early in the library
序列一開始讀的時候,由於固定位置的組成序列會較複雜而會無法做出精確的base call,這種情形在Per Base Sequence Content裡面也會觀察到開頭的content較混亂的情形。
//敘述不太好不易懂,回頭想到再寫。
- general loss of quality
Sequence Length Distribution
說明:顯示read讀長的分布。
- X軸:讀長。
- Y軸:某個讀長的read數量。
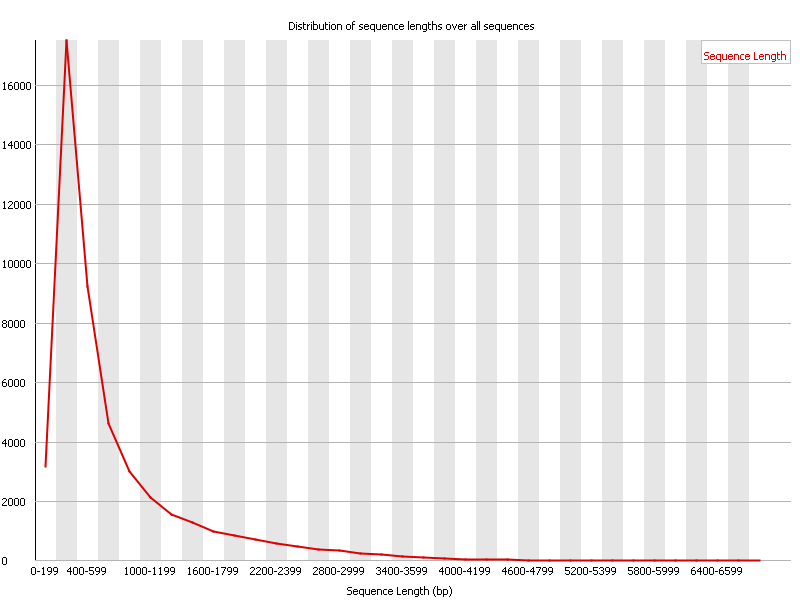
異常:
- Warning:代表read的長度不均(猜SE過大)。
- Failure:有一個以上的read長度是0。
- Common reasons:現在許多平台的讀長不均是正常現象,可以忽略這個module的報錯。
Sequence Duplication Levels
說明:顯示一模一樣的序列出現在不同read的頻率。
一個良好的library理論上每個序列只會出現一次(即使都mapping到genome的同一個site的附近,fragment的時候因為斷點是隨機的因此會產生的序列彼此之間也會有shift)。
- Low Duplication Level:high coverage。
- Higher Duplication Level:可能有一些enrichment bias (ex:pcr over amplification)。
- X軸:重複次數(>10的部分非等距)。
- Y軸:某個重複次數的read種類。
- 藍色線:deduplicate之前的樣子。
- 紅色線:deduplicate之後的樣子。
//既然都deduplicate了為何還有重複的,原理未知?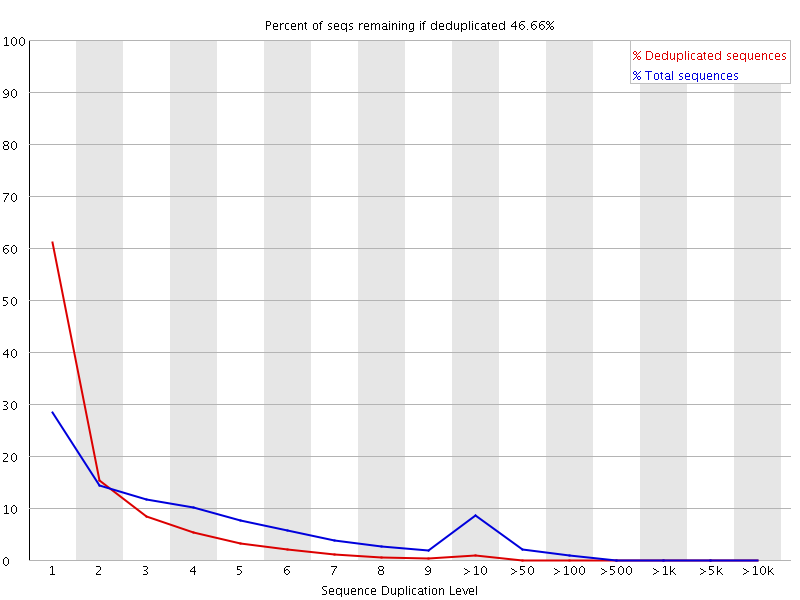
為了節省記憶體使用,fastqc只會分析每個檔案的前100,000個序列,但這樣的樣本數已經足以做Duplication level的判斷。
由於這個module需要判斷兩條read每個site的序列都完全一樣才會成是duplicate,而長一些的序列較末端的定序品質不穩定,如果拿整條序列來比就無法判斷為duplicate,因此module會將讀長超過75bp以上的read切成50bp來做比較,即使如此讀長較長的序列還是較容易誤判為不同條而增加diversity,而傾向於低估duplication level。
specific enrichments of subsets、presence of low complexity contaminants會增加duplication level拉平曲線
異常:
- Warning: non-unique sequences > 20%。
- Failure:non-unique sequences > 50%。。
- Common reasons:。
Overrepresented sequences
Adapter Content
Kmer Content
Reference
Babraham bioinformatics
我們的基因體時代 OUR "GENE"RATION-RNAseq’s 資料前處理:Quality Control